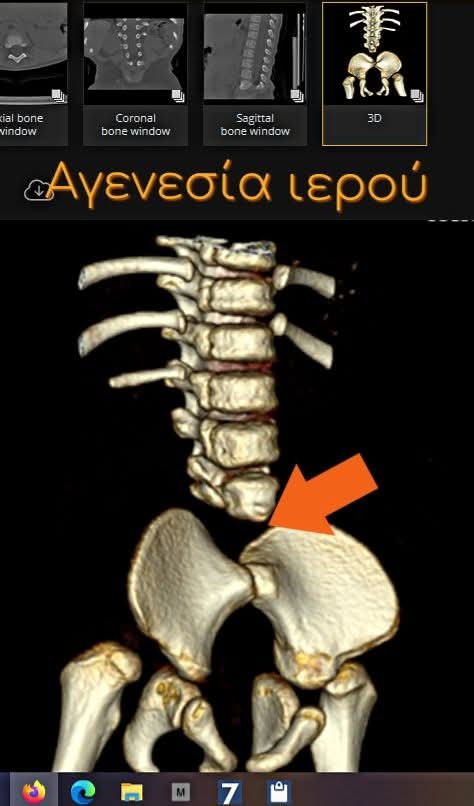
Ο Κρυφός Ραχιαίος Δισχιδισμός (διεθνώς γνωστός ως Spina Bifida Occulta ή λανθάνουσα δισχιδής ράχη) είναι η πιο ήπια και συχνή μορφή δισχιδούς ράχης. Χαρακτηρίζεται από ένα μικρό έλλειμμα στη σύγκλιση των σπονδυλικών τόξων, το οποίο καλύπτεται πλήρως από το δέρμα, ενώ ο νωτιαίος μυελός και οι νευρικές ρίζες παραμένουν συνήθως άθικτα μέσα στον σπονδυλικό σωλήνα. Επηρεάζει το 10% έως 20% του γενικού πληθυσμού και οι περισσότεροι άνθρωποι δεν γνωρίζουν ποτέ ότι το έχουν.
Ο κρυφός ραχιαίος δισχιδισμός (Occult Spinal Dysraphism – OSD) αποτελεί μια ομάδα συγγενών ανωμαλιών της σπονδυλικής στήλης, όπου τα οστά της σπονδυλικής στήλης δεν κλείνουν πλήρως γύρω από τον νωτιαίο μυελό, αλλά η βλάβη καλύπτεται από δέρμα. Σε αντίθεση με τον ανοιχτό δισχιδισμό, ο νευρικός ιστός δεν είναι εκτεθειμένος στο εξωτερικό περιβάλλον. Ωστόσο, μπορεί να συνυπάρχουν υποκείμενες παθολογίες όπως το σύνδρομο καθηλωμένου μυελού (tethered cord syndrome), λιπώματα ή δερμοειδείς κύστεις, που μπορεί να προκαλέσουν προοδευτική νευρολογική βλάβη καθώς το παιδί μεγαλώνει. Η απλή μορφή της κρυφής δισχιδούς ράχης (spina bifida occulta) είναι εξαιρετικά συχνή, εμφανιζόμενη στο 10-20% του γενικού πληθυσμού, και συχνά είναι ασυμπτωματική. Ωστόσο, ο κλινικά σημαντικός κρυφός ραχιαίος δισχιδισμός, που συνοδεύεται από δερματικά στίγματα και υποκείμενες νευρολογικές ανωμαλίες που απαιτούν παρέμβαση, είναι λιγότερο συχνός αλλά κλινικά κρίσιμος για την πρόληψη μόνιμων βλαβών.
Στη συντριπτική πλειονότητα των περιπτώσεων, η πάθηση είναι ασυμπτωματική και ανακαλύπτεται τυχαία κατά τη διάρκεια απεικονιστικού ελέγχου (π.χ. ακτινογραφία, μαγνητική τομογραφία) για άλλο λόγο. Ωστόσο, στις περισσότερες περιπτώσεις, τα πρώτα σημάδια είναι ορατά στο δέρμα της οσφυϊκής περιοχής (χαμηλά στην πλάτη) αμέσως μετά τη γέννηση. Αυτά τα “δερματικά στίγματα” περιλαμβάνουν:
Υπερτρίχωση (μια τούφα από τρίχες στην περιοχή της σπονδυλικής στήλης).
Εμβύθιση του δέρματος (λακκάκι) ή δερματικός κόλπος (dimple).
Αιμαγγείωμα (κόκκινο σημάδι ή “φράουλα” στο δέρμα).
Υποδόριο λίπωμα (μαλακό εξόγκωμα κάτω από το δέρμα).
Αποχρωματισμός του δέρματος (υπομελάγχρωση ή υπερμελάγχρωση).
Καθώς το παιδί αναπτύσσεται, μπορεί να εμφανιστούν συμπτώματα που οφείλονται στην καθήλωση του νωτιαίου μυελού (tethering), η οποία εμποδίζει την ελεύθερη κίνηση του μυελού εντός του σωλήνα:
Δυσκολία στη βάδιση: Ασυμμετρία στο περπάτημα ή συχνές πτώσεις.
Πόνος: Πόνος στη μέση ή στα πόδια, ο οποίος μπορεί να επιδεινώνεται με τη δραστηριότητα.
Ορθοπεδικές παραμορφώσεις: Πόδια που στρέφονται προς τα μέσα ή παραμορφώσεις των άκρων ποδών (κοιλοποδία).
Διαταραχές σφιγκτήρων: Απώλεια ελέγχου της ουροδόχου κύστης (ενούρηση) ή του εντέρου. Η εμφάνιση συμπτωμάτων που αφορούν τη λειτουργία της κύστης και του εντέρου είναι ενδεικτική αρχόμενης βλάβης στα νεύρα και απαιτεί άμεση αξιολόγηση.
Ο κρυφός ραχιαίος δισχιδισμός συχνά συνυπάρχει με:
Σκολίωση ή Κύφωση (παραμορφώσεις της σπονδυλικής στήλης).
Συγγενές εξάρθρημα του ισχίου.
Ανωμαλίες του ουροποιητικού συστήματος (π.χ. παλινδρόμηση ούρων).
Σύνδρομο VACTERL (συνδυασμός ανωμαλιών που επηρεάζουν σπονδύλους, πρωκτό, καρδιά, τραχεία, οισοφάγο, νεφρά και άκρα).
Η πιο σημαντική επιπλοκή είναι το σύνδρομο καθηλωμένου μυελού (tethered cord). Καθώς η σπονδυλική στήλη μακραίνει ταχύτερα από τον νωτιαίο μυελό, ο μυελός τραβιέται προς τα κάτω, προκαλώντας ισχαιμία και βλάβη στους νευρώνες. Αυτό οδηγεί σε μόνιμη ακράτεια, σεξουαλική δυσλειτουργία στην ενήλικη ζωή και κινητική αναπηρία.
Για την απλή, ασυμπτωματική μορφή (Spina Bifida Occulta) δεν απαιτείται καμία απολύτως θεραπεία ή περιορισμός δραστηριοτήτων. Χειρουργική αντιμετώπιση απαιτείται μόνο εάν διαπιστωθεί καθήλωση του μυελού (tethered cord) ή άλλη συνοδός δυσπλαστική μάζα που πιέζει τα νεύρα, με σκοπό την αποσυμπίεση.